
Independent Teams
A space managed by WorkflowHub administrators for teams that don't want/need to manage their own space.
Web page: Not specified
Funding details: Related items
Teams: CO2MICS Lab
Organizations: Biomedical Research Foundation (BRFAA) of the Academy of Athens
Teams: Values Shape Community
Organizations: Values Shape Community
Hi, I'm Paul, a dedicated academic writer and researcher with a passion for crafting insightful essays. I've been helping students elevate their academic work through clear, well-researched writing. My expertise spans various subjects, with a special focus on business ethics and philosophical analysis.
At AllEssay, I share carefully curated resources to help students develop stronger arguments and ethical frameworks in their writing. You'll find one of my most useful guides on crafting definition ...
Teams: RTC Bioinformatics
Organizations: Radboud University Medical Center

Teams: Medvedeva Lab
Organizations: Moscow Institute of Physics and Technology
Teams: MLme: Machine Learning Made Easy
Organizations: University of Bern
Teams: IBISBA Workflows
Organizations: University of Saskatchewan
Teams: Mr.
Organizations: Univrsity of kufa

Teams: UX trial team
Organizations: The University of Manchester
Teams: IBISBA Workflows
Organizations: The University of Manchester
Teams: ELIXIR Training, ELIXIR Tools platform, ELIXIR Belgium
Organizations: VIB, ELIXIR Belgium
 https://orcid.org/0000-0002-6675-3836
https://orcid.org/0000-0002-6675-3836
Teams: SKM3
Organizations: The Open University

Space: Independent Teams
Public web page: https://fairagro.net/
Space: Independent Teams
Public web page: Not specified
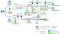
SyBR: a scalable Snakemake workflow for synteny inference, evolutionary breakpoint detection, and functional enrichment analysis
Space: Independent Teams
Public web page: https://sybr.igib.res.in/
a team for BH2025 project 30
Space: Independent Teams
Public web page: https://github.com/elixir-europe/biohackathon-projects-2025/blob/main/30.md
Start date: 3rd Nov 2025

Research Programme Smart Data Technologies (SDT) at Leibniz Institute for Plasma Science and Technology (INP) in Greifswald, Germany.
Space: Independent Teams
Public web page: https://www.inp-greifswald.de/en/research/smart-data-technologies
Workflows developed by EMBL Hentze Group bioinformatics team for NGS data processing
Space: Independent Teams
Public web page: https://www.embl.org/groups/hentze/
Space: Independent Teams
Public web page: https://compbio.ca
The research group focuses on chemical data in the context of biological systems. The investigated areas include structural bioinformatics of proteins and the application of computational methods of molecular modeling for designing the structural activity of new small molecules for specific biological effects.Another focus of the group is the development and operation of chemical databases and tools. The MolMeDB database collects physicochemical data on interactions of small molecules with membranes ...
Space: Independent Teams
Public web page: https://www.kfc.upol.cz/en/
Space: Independent Teams
Public web page: https://conesalab.org

The team collects the workflow and workflow-related training activities related to ELIXIR Norway, run by ELIXIR Norway or affiliated to ELIXIR Norway.
Space: Independent Teams
Public web page: https://www.elixir.no/
Space: Independent Teams
Public web page: Not specified
Space: Independent Teams
Public web page: Not specified
Exeter Genetics team
Space: Independent Teams
Public web page: https://github.com/ExeterGenetics/
Space: Independent Teams
Public web page: Not specified
Space: Independent Teams
Public web page: Not specified
Automated pipeline for generating residue-level lipid contact annotations from PDB structures. The workflow queries RCSB PDB for structures containing lipid chemical components (117 recognized codes), downloads original PDB files, calculates all-atom heavy-atom contacts at 4.0 Angstrom cutoff, clusters sequences at 30% identity using MMseqs2, and partitions into train/validation/test splits. Produces a complete dataset of 4,704 proteins with 8,055,325 annotated residues.
Space: Independent Teams
Public web page: https://github.com/omagebright/MPLID
This is for testing purposes only
Space: Independent Teams
Public web page: Not specified
Space: Independent Teams
Public web page: Not specified
Space: Independent Teams
Public web page: Not specified

Our research group Methods and Algorithms for Bioinformatics belongs to French National Center for Scientific Research (CNRS) and is hosted by the Computer Science Dpt of University of Montpellier (in the LIRMM joint unit UMR 5506). We offer interactive web services, software, and programming libraries for bioinformatics on the ATGC bioinformatic platform. Well known software from our group are for instance ...
Space: Independent Teams
Public web page: http://www.atgc-montpellier.fr/
ROR ID: Not specified
Department: Not specified
Country: Not specified
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country: Not specified
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country:  Poland
Poland
City: Poznan
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country: Not specified
City: Not specified
Web page: Not specified
ROR ID: https://ror.org/0066zpp98
Department: Not specified
Country:  China
China
City: Shenzhen
Web page: http://www.agis.org.cn/
ROR ID: Not specified
Department: Not specified
Country:  Ethiopia
Ethiopia
City: Aksum
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country:  Germany
Germany
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country: Not specified
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country:  Australia
Australia
City: Not specified
Web page: Not specified

ROR ID: https://ror.org/05sd8tv96
Department: Not specified
Country:  Spain
Spain
City: Barcelona
Web page: https://www.bsc.es/
ROR ID: Not specified
Department: Not specified
Country:  Canada
Canada
City: Vancouver
Web page: https://bcgsc.ca/
ROR ID: Not specified
Department: Not specified
Country:  Belgium
Belgium
City: Liege
Web page: https://bccm.belspo.be/about-us/bccm-ulc
ROR ID: Not specified
Department: Not specified
Country: Germany
City: Berlin
Web page: https://bihealth.org
ROR ID: Not specified
Department: Not specified
Country: Not specified
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country: Not specified
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country: Not specified
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country: Not specified
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country: Germany
City: Bielefeld
Web page: Not specified
ROR ID: Not specified
Department: Not specified
Country:  France
France
City: Not specified
Web page: Not specified
ROR ID: Not specified
Department: Bioinformatics and data-analysis
Country: Not specified
City: Oslo
Web page: https://www.vetinst.no
This dataset is an RO-Crate representation of an execution of the tissue/tumor prediction workflow for digital pathology from crs4/deephealth-pipelines. It follows the Provenance Run Crate profile. The workflow has been run with cwltool, using the --provenance option to generate a CWLProv RO bundle, and then converted to an RO-Crate using runcrate. The input dataset is Mirax2-Fluorescence-2 by Yves Sucaet, from the MIRAX test data.
Creator: Simone Leo
Submitter: Stian Soiland-Reyes


Keçeci Numbers: Keçeci Sayıları
Description / Açıklama
Keçeci Numbers (Keçeci Sayıları): Keçeci Numbers; An Exploration of a Dynamic Sequence Across Diverse Number Sets: This work introduces a novel numerical sequence concept termed "Keçeci Numbers." Keçeci Numbers are a dynamic sequence generated through an iterative process, originating from a specific starting value and an increment value. In each iteration, the increment value is added to the current value, and this "added value" is recorded ...
 Download
Downloadadnus (AdNuS): Advanced Number Systems
adnus is a Python library that provides an implementation of various advanced number systems. This library is designed for mathematicians, researchers, and developers who need to work with number systems beyond the standard real and complex numbers. Features
Harmonic and Oresme Sequences: Functions to generate harmonic numbers and Oresme sequences. Bicomplex Numbers: A class for bicomplex numbers with full arithmetic support. Neutrosophic Numbers: Classes ...
KececiLayout
Kececi Layout (Keçeci Yerleşimi): A deterministic graph layout algorithm designed for visualizing linear or sequential structures with a characteristic "zig-zag" or "serpentine" pattern.
Python implementation of the Keçeci layout algorithm for graph visualization. Description / Açıklama
This algorithm arranges nodes sequentially along a primary axis and offsets them alternately along a secondary axis. It's particularly useful for path graphs, chains, or showing progression.
Bu ...
Oresmej: Oresme Jax
Oresme numbers refer to the sums related to the harmonic series. Türkçe Tanım:
Oresme Sayıları, 14. yüzyılda Nicole Oresme tarafından incelenen matematiksel serilerdir. Oresme sayıları harmonik seriye ait toplamları ifade eder. İki türü vardır:
( \frac{n}{2^n} ) serisi (Oresme'nin orijinal çalışması), Harmonik sayılar (( H_n = 1 + \frac{1}{2} + \cdots + \frac{1}{n} )). Bu sayılar, analiz ve sayı teorisinde önemli rol oynar.
English Definition:
Oresme Numbers are mathematical ...
Türkçe Tanım:
Oresme Sayıları, 14. yüzyılda Nicole Oresme tarafından incelenen matematiksel serilerdir. Oresme sayıları harmonik seriye ait toplamları ifade eder. İki türü vardır:
( \frac{n}{2^n} ) serisi (Oresme'nin orijinal çalışması), Harmonik sayılar (( H_n = 1 + \frac{1}{2} + \cdots + \frac{1}{n} )). Bu sayılar, analiz ve sayı teorisinde önemli rol oynar.
English Definition:
Oresme Numbers are mathematical series studied by Nicole Oresme in the 14th century. Oresme numbers refer to the sums ...
Keçeci Fractals: Keçeci Fraktals
Keçeci Circle Fractal: Keçeci-style circle fractal.
Description / Açıklama
Keçeci Circle Fractal: Keçeci-style circle fractal.:
This module provides two primary functionalities for generating Keçeci Fractals:
kececifractals_circle(): Generates general-purpose, aesthetic, and randomly colored circular fractals. visualize_qec_fractal(): Generates fractals customized for modeling the (version >= 0.1.1) concept of Quantum Error Correction (QEC) codes. Stratum ...
Keçeci Binomial Squares (Keçeci Binom Kareleri): Keçeci's Arithmetical Square (Keçeci Aritmetik Karesi, Keçeci'nin Aritmetik Karesi)
Description / Açıklama
Keçeci Binomial Squares (Keçeci Binom Kareleri): Keçeci's Arithmetical Square (Keçeci Aritmetik Karesi, Keçeci'nin Aritmetik Karesi):
Keçeci Binomial Squares (Keçeci Binom Kareleri): The Keçeci Binomial Square is a series of binomial coefficients forming a square region within Khayyam (مثلث خیام), Pascal, Binomial Triangle, selected from a ...
grikod2 (Gri Kod, Gray Code)
A Python library for converting binary numbers to Gray Code with ease. Tanım (Türkçe)
Gri Kod: grikod2 İkili sayıları Gri Koda çevirir. Description (English)
Gri Kod: grikod2 converts binary numbers to Gray Code. Kurulum (Türkçe) / Installation (English) Python ile Kurulum / Install with pip, conda, mamba
pip install grikod2 -U python -m pip install -U grikod2 conda install bilgi::grikod2 -y mamba install bilgi::grikod2 -y
- pip uninstall grikod2 -y
- pip install ...
Grikod (Gri Kod, Gray Code)
Tanım (Türkçe) Gri Kod: Grikod İkili sayıları Gri Koda çevirir.
Description (English) Gri Kod: Grikod converts binary numbers to Gray Code.
Kurulum (Türkçe) / Installation (English) Python ile Kurulum / Install with pip, conda, mamba pip install grikod -U python -m pip install -U grikod conda install bilgi::grikod -y mamba install bilgi::grikod -y
- pip uninstall grikod -y
- pip install -U grikod
- python -m pip install -U grikod PyPI
Test Kurulumu / Test Installation ...
This is metadata of human trafficking research conducted by Daniel Tesfa in Tigray and Addis Ababa between October 2024 and March 2025.
Creator: Daniel Tesfa
Submitter: Daniel Tesfa
Download all genome from https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide with filter host:viridiplantae and Refseq on.
Creator: johan Rollin
Submitter: johan Rollin
Creator: Felix Shaw
Submitter: Felix Shaw


Abstract (Expand)
Authors: Alexandre Flageul, Edouard Hirchaud, Céline Courtillon, Flora Carnet, Paul Brown, Béatrice Grasland, Fabrice Touzain
Date Published: 17th Oct 2025
Publication Type: Journal Article
Abstract
Authors: Alexandre Flageul, Edouard Hirchaud, Céline Courtillon, Flora Carnet, Paul Brown, Béatrice Grasland, Fabrice Touzain
Date Published: 2025
Publication Type: Other
Abstract (Expand)
Author: Fabrice Touzain
Date Published: 2025
Publication Type: Dataset







Abstract (Expand)
Authors: Simone Leo, Michael R. Crusoe, Laura Rodríguez-Navas, Raül Sirvent, Alexander Kanitz, Paul De Geest, Rudolf Wittner, Luca Pireddu, Daniel Garijo, José M. Fernández, Iacopo Colonnelli, Matej Gallo, Tazro Ohta, Hirotaka Suetake, Salvador Capella-Gutierrez, Renske de Wit, Bruno P. Kinoshita, Stian Soiland-Reyes
Date Published: 10th Sep 2024
Publication Type: Journal Article


Abstract
Authors: João Vitor F. Cavalcante, Iara Dantas de Souza, Diego A. A. Morais, Rodrigo J. S. Dalmolin
Date Published: 27th Aug 2024
Publication Type: Conference Paper
Abstract (Expand)
Authors: W.T.K. Maassen, L.F. Johansson, B. Charbon, D. Hendriksen, S. van den Hoek, M.K. Slofstra, R. Mulder, M.T. Meems-Veldhuis, R. Sietsma, H.H. Lemmink, C.C. van Diemen, M.E. van Gijn, M.A. Swertz, K.J. van der Velde
Date Published: 15th Apr 2024
Publication Type: Preprint


Abstract (Expand)
Authors: Mahnoor Zulfiqar, Michael R. Crusoe, Birgitta König-Ries, Christoph Steinbeck, Kristian Peters, Luiz Gadelha
Date Published: 1st Feb 2024
Publication Type: Journal Article
Abstract (Expand)
Authors: Mahnoor Zulfiqar, Michael R. Crusoe, Birgitta König-Ries, Christoph Steinbeck, Kristian Peters, Luiz Gadelha
Date Published: 21st Dec 2023
Publication Type: Journal Article
Abstract (Expand)
Authors: Thibault Poinsignon, Mélina Gallopin, Pierre Grognet, Fabienne Malagnac, Gaëlle Lelandais, Pierre Poulain
Date Published: 1st Dec 2023
Publication Type: Journal Article


Abstract (Expand)
Author: Yasmmin Martins
Date Published: 28th Sep 2023
Publication Type: Journal Article
Abstract (Expand)
Authors: Yasmmin Côrtes Martins, Ronaldo Francisco da Silva
Date Published: 27th Sep 2023
Publication Type: Journal Article
Abstract (Expand)
Authors: Yasmmin Martins, Ronaldo Francisco da Silva
Date Published: 22nd Jun 2023
Publication Type: Journal Article
Abstract (Expand)
Author: Yasmmin C Martins
Date Published: 7th Jun 2023
Publication Type: Journal Article
Abstract (Expand)
Authors: Yasmmin Côrtes Martins, Artur Ziviani, Maiana de Oliveira Cerqueira e Costa, Maria Cláudia Reis Cavalcanti, Marisa Fabiana Nicolás, Ana Tereza Ribeiro de Vasconcelos
Date Published: 2023
Publication Type: Journal Article


Abstract (Expand)
Authors: Rafael Terra, Kary Ocaña, Carla Osthoff, Lucas Cruz, Philippe Navaux, Diego Carvalho
Date Published: 19th Oct 2022
Publication Type: Conference Paper
Abstract (Expand)
Authors: Andrzej Oleksa, Eliza Căuia, Adrian Siceanu, Zlatko Puškadija, Marin Kovačić, M. Alice Pinto, Pedro João Rodrigues, Fani Hatjina, Leonidas Charistos, Maria Bouga, Janez Prešern, Irfan Kandemir, Slađan Rašić, Szilvia Kusza, Adam Tofilski
Date Published: 1st Oct 2022
Publication Type: Journal Article
Abstract (Expand)
Authors: Rafael Terra, Kary Ocaña, Carla Osthoff, Diego Carvalho
Date Published: 18th Feb 2022
Publication Type: Master's Thesis
Abstract (Expand)
Authors: Yasmmin Côrtes Martins, Artur Ziviani, Marisa Fabiana Nicolás, Ana Tereza Ribeiro de Vasconcelos
Date Published: 6th Sep 2021
Publication Type: Journal Article
Abstract (Expand)
Authors: Rafael Terra, Micaella Coelho, Lucas Cruz, Marco Garcia-Zapata, Luiz Gadelha, Carla Osthoff, Diego Carvalho, Kary Ocaña
Date Published: 18th Jul 2021
Publication Type: Conference Paper

Abstract (Expand)
Authors: Michael R. Crusoe, Sanne Abeln, Alexandru Iosup, Peter Amstutz, John Chilton, Nebojša Tijanić, Hervé Ménager, Stian Soiland-Reyes, Carole Goble
Date Published: 14th May 2021
Publication Type: Preprint
This presentation was given at the UK DARIAH day at Edinburgh.
Creator: karina-rodriguez
Submitter: karina-rodriguez


This document provides guidance for understanding and implementing the set of workflows included in this collection. It outlines best practices and recommendations to ensure successful setup and execution of automated and real-time image processing pipelines.
Creator: Daniel Marchan
Submitter: Daniel Marchan
Guidelines for using Scipion in the context of on-the-fly image processing at cryo-EM facilities. This document is divided into three main sections:
-
On-the-fly processing: Monitoring data acquisition in real time.
-
Processing workflows and templates: Building familiarity with creating and customizing workflows and templates in Scipion.
-
Scipion with queue systems: Demonstrating Scipion’s behavior and integration with SLURM-based queueing systems.
Creator: Daniel Marchan
Submitter: Daniel Marchan
This document provides a detailed explanation of all the workflows, including their functionalities, problems they address, advantages, disadvantages, implementation requirements, and open points for future versions.
Creator: Daniel Marchan
Submitter: Daniel Marchan
In the age of high-throughput data, computational workflows have made data processing tasks flexible, manageable, and automated. To administer different computational activities in a workflow, different workflow management systems (WMS) are used that necessitate a sophisticated level of standardisation. Standardisation and reproducibility can be achieved by using standard formats for specifying workflows, such as Common Workflow Language (CWL), and provenance gathering with the standard W3C PROV ...
Creator: Mahnoor Zulfiqar
Submitter: Mahnoor Zulfiqar
This is human trafficking vecabulory that explains how terms are used to explore the human trafficking context in Ethiopia.
ConceptScheme URI https://workflowhub.eu/events/11 PREFIX vocab https://workflowhub.eu/events/11 dct:title Human Trafficking Vocabulary dct:description Welcome to Human Trafficking Vocabulary dct:creater https://orcid.org/0000-0002-5115-0231
URI skos:prefLabel@en skos:altLabel@en skos:definition@en skos:narrower(separator=",") skos:notation@en vocab:type_of_data Data Type ...
Start Date: 22nd Apr 2025 (22nd Apr 2025 (Africa/Nairobi))
End Date: 22nd Apr 2025 (22nd Apr 2025 (Africa/Nairobi))
Event type: Not specified
Weyl semimetals (WSMs) have emerged as one of the most groundbreaking discoveries in condensed matter physics in recent years, deepening our fundamental scientific understanding and opening new horizons for future technologies. These materials are named after Hermann Weyl, who in 1929 predicted Weyl fermions – massless, chiral relativistic particles. WSMs, as low-energy excitations of these fermions in solid-state systems, possess special points in their electronic band structure where bands cross ...
Solving the Hamiltonian Problem in Graph Theory Education with Z3 and the Keçeci Layout
Mehmet Keçeci ORCID: https://orcid.org/0000-0001-9937-9839
Received: 08.21.2025
Abstract: Whilst graph theory constitutes a cornerstone of computer science and mathematics education, the abstract nature of NP-complete problems, such as the Hamiltonian cycle problem, presents a significant conceptual challenge for students. An intuitive grasp of such problems often requires the analytical inspection of complex ...
Graf teorisi, bilgisayar bilimleri ve matematik eğitiminin temel taşlarından birini oluşturmakla birlikte, Hamilton döngüsü gibi NP-tam problemlerin soyut doğası, öğrenciler için önemli bir anlama zorluğu teşkil etmektedir. Bu tür problemlerin sezgisel olarak kavranması, genellikle karmaşık ve standart dışı graf yapılarının analitik olarak incelenmesini gerektirir. Geleneksel öğretim metotları, bu karmaşıklığı etkili bir şekilde aktarmada yetersiz kalabilmekte ve öğrencilerin konuya olan ilgisini ...
Hilbert spaces provide the fundamental mathematical framework for describing quantum mechanical systems. Their structure, characterized by an inner product and completeness, allows for the representation of quantum states as vectors and physical observables as self-adjoint operators. Key quantum phenomena such as superposition and entanglement find natural expression within this formalism. Superposition, where a quantum system can exist in multiple states simultaneously, is represented by linear ...
The development of quantum computers represents one of the most exciting and challenging endeavours in modern science. Progress in this field, particularly with the advent of the second quantum revolution, necessitates a profound interplay between diverse disciplines such as materials science, condensed matter physics, and quantum information theory. The primary aim of this work is to explore the potential of layered quantum structures containing exotic particles—Weyl and Majorana fermions—to ...
Keçeci Varsayımı'nın Hesaplanabilirliği: Sonlu Adımda Kararlı Yapıya Yakınsama Sorunu
Mehmet Keçeci ORCID: https://orcid.org/0000-0001-9937-9839, Independent Researcher
Received: 03.08.2025
Abstract:
Bu çalışma, "Keçeci Varsayımı" olarak adlandırılan yeni bir matematiksel hipotezi sunar ve bu varsayımın, klasik Collatz Varsayımı'nın çok daha genel bir çerçevede formüle edilmiş hali olabileceğini ileri sürer. Keçeci Varsayımı, belirli bir yinelemeli süreç boyunca, herhangi bir başlangıç değerinin ...
Bu çalışma, harmonik serilerin hesaplanmasında geleneksel saf Python tabanlı bir yaklaşım ile modern JAX kütüphanesine dayalı bir yaklaşımın performans, ölçeklenebilirlik ve verimlilik açısından karşılaştırılmasını sunmaktadır. Harmonik seri ve matematiksel analizde önemli bir yere sahip olan ıraksak bir seridir. Hesaplamalı matematikte bu tür serilerin etkin bir şekilde değerlendirilmesi, özellikle büyük veri ve yüksek hassasiyet gerektiren uygulamalarda kritik öneme sahiptir. Geleneksel saf ...
The Limits of Python in Computational Mathematics and Their Extension with JAX: An Application on Harmonic Numbers
Mehmet Keçeci ORCID: https://orcid.org/0000-0001-9937-9839, Independent Researcher
Received: 29.07.2025
Abstract:
This study presents a comparative analysis of a traditional, pure Python-based approach and a modern, JAX library-based approach for calculating the harmonic series, focusing on performance, scalability, and efficiency. The harmonic series is a divergent series of significant ...
This study extends the domain of Keçeci Numbers, a unique class of sequences generated by a distinctive algorithm, beyond standard number systems into advanced mathematical structures such as neutrosophic and hyperreal numbers. Keçeci Numbers are sequences produced via a recursive algorithm based on a starting value and an incremental scalar, governed by rules of divisibility and primality. The primary objective of this paper is to investigate the behaviour of this deterministic algorithm within ...
Bu çalışma, özgün bir algoritmik yapıya sahip olan Keçeci Sayı dizilerinin tanım kümesini, standart sayı sistemlerinin ötesine taşıyarak nötrosofik ve hipergerçek sayılar gibi ileri düzey matematiksel yapılarla genişletmektedir. Keçeci Sayıları, temel olarak bir başlangıç değeri ve bir artış skalerine dayanan, bölünebilirlik ve asallık testlerine bağlı özyinelemeli bir algoritma ile üretilen dizilerdir. Bu çalışmanın temel amacı, bu deterministik algoritmanın, belirsizlik (nötrosofik) ve sonsuz ...
This study presents a comparative analysis of static and dynamic number sequences, using the classical Oresme numbers and the novel Keçeci numbers, developed by Mehmet Keçeci, as primary case studies. Static sequences are characterized by a fixed, predictable recurrence relation. The Oresme numbers—the partial sums of the harmonic series (Η_n=∑(k=1)^n 1/k)—exemplify this category. Their generation follows a simple, deterministic rule (Η_n= Η(n-1)+1/n), and their predictable divergence, proven ...
Nodal-line semimetals constitute a fascinating subclass within the family of topological materials in condensed matter physics. These unique materials are characterized by the intersection of their electronic energy bands along one or more closed or open lines within the Brillouin Zone, rather than at isolated points. These lines of intersection are termed "nodal lines," and along these lines, electrons can behave as if they were massless, leading to extraordinary electronic properties. The ...
Beyond Topology: Deterministic and Order-Preserving Graph Visualization with the Keçeci Layout
Mehmet Keçeci1 1ORCID : https://orcid.org/0000-0001-9937-9839, İstanbul, Türkiye
Received: 03.07.2025
Özet/Abstract:
The visualization of scientific data is a fundamental step in uncovering hidden patterns and relationships within complex systems. While conventional force-directed graph layout algorithms (e.g., spring layout) are effective at displaying the general topology and clustering tendencies of ...
This study comprehensively examines the profound technological and methodological synergies existing between observations made via interferometric detectors such as LIGO and Virgo, which have revolutionized the field of gravitational wave (GW) astrophysics, and the rapidly advancing quantum computing (QC) technologies. As both disciplines aim to perform measurements pushing the limits of precision, the effective control and mitigation of environmental and quantum-originated noise pose a critical ...
Many systems encountered in nature and engineering exhibit complex and hierarchical geometric structures. Fractal geometry provides a powerful tool for understanding and modeling these structures. However, existing deterministic circle packing fractals, such as the Apollonian gasket, often adhere to fixed geometric rules and may fall short in accurately reflecting the diversity of observed structures. Addressing the need for greater flexibility in modeling physical and mathematical systems, this ...
Keçeci Deterministic Zigzag Layout (Keçeci Zigzag Layout Algorithm, Keçeci Layout) is a deterministic node layout algorithm designed for graph visualization in Python. Its primary purpose is to position the nodes of a graph in a predefined, sequential, and repeatable manner. The algorithm processes nodes sequentially, placing them along a user-defined primary axis (e.g., top-down or left-to-right) while applying an offset on the secondary axis in a zigzag pattern. This zigzag pattern helps prevent ...
Creator: Liang Cheng
Submitter: Liang Cheng
This workflow is part of the EJP RD case study on CAKUT published here: Bayjanov, J.R., Doornbos, C., Ozisik, O. et al. Integrative analysis of multi-omics data reveals importance of collagen and the PI3K AKT signalling pathway in CAKUT. Sci Rep 14, 20731 (2024). https://doi.org/10.1038/s41598-024-71721-8
Creator: Juma Bayjan
Submitter: Juma Bayjan


Creator: Jasper Koehorst
Submitter: Jasper Koehorst
Protein domains can be viewed as building blocks, essential for understanding structure-function relationships in proteins. However, each domain database classifies protein domains using its own methodology. Thus, in many cases, boundaries between different domains or families differ from one domain database to the other, raising the question of domain definition and enumeration. The answer to this question cannot be found in a single database. Rather, expert integration and curation of various ...
Creators: Hrishikesh Dhondge, Isaure Chauvot de Beauchêne, Marie-Dominique Devignes
Submitter: Hrishikesh Dhondge
Bioimaging Data Management Workflow for Plasma Medicine
This repository contains the Jupyter notebook implementation of a bioimaging data management workflow for plasma medicine (see preprint and poster). The workflow is implemented as a structured pipeline integrating open-source tools, including OMERO for image data management, eLabFTW ...
Type: Jupyter
Creators: Mohsen Ahmadi , Robert Wagner , Sander Bekeschus , Markus M. Becker
Submitter: Mohsen Ahmadi

EXCON (v2.3.2)
A Nextflow pipeline for gene family EXpansion and CONtraction analysis across multiple species using CAFE5.
Given a set of genome assemblies and annotations, EXCON builds orthogroups with OrthoFinder, fits and compares multiple CAFE models to identify gene families evolving at significantly different rates, and automatically selects the best-fitting model for downstream analysis. Optionally, GO enrichment analysis can be run on expanded and contracted gene families, and ...
Phorager
Prophage Analysis Pipeline
A comprehensive pipeline for bacterial genome quality control, prophage detection, and prophage characterization
Authors:
- Xena Dyball
- James Docherty
- Alise Ponsero
- Ryan Cook
Citation: [Paper Citation or Preprint Link]
Overview
Phorager provides an integrated workflow for:
- Bacterial genome quality control - CheckM2 assessment, filtering, and dRep dereplication
- Prophage detection - GenoMAD and ...
CLIP-seq Workflow
A Nextflow workflow for end-to-end processing of CLIP-seq data, supporting multiple CLIP protocols.
Overview
Starting from raw FASTQ files (or un-demultiplexed iCLIP data), the workflow processes reads through quality control, adapter trimming, rRNA removal, genome alignment, and UMI deduplication, then runs shoji to extract crosslink sites and produce per-sample and combined count matrices ready for differential binding analysis (see ...

Petrisnake: A secondary analysis pipeline for PETRI-seq data.
This is a Snakemake pipeline for the secondary computational analysis of single cell RNA-seq data from the PETRI-seq protocol (https://www.nature.com/articles/s41564-020-0729-6 and https://www.nature.com/articles/s41586-024-08124-2), this is: From the input FASTQ files, this workflow constructs a gene count table showing the expression of each gene in each cell. Petrisnake is available on WorkflowHub (https://workflowhub.eu/workflows/2081). ...
Automated screening monitoring workflow designed to provide real-time feedback to the microscope during data acquisition. The pipeline processes movies on-the-fly, performing motion correction, maxshift analysis, CTF estimation and consensus validation, as well as micrograph quality assessment through AI-based categorization. The workflow also includes particle picking, automatic box size estimation, particle extraction, streaming 2D classification, and 2D class quality assessment. Results are ...
This workflow performs a basic Virtual Drug Screening Pipeline. THe workflows uses the following protocols: 1.a) Import receptor: Imports the atomic structure with PDB code 4ERF. 1.b) Import small molecules: Imports a set of 4 molecules from local files. 2.a) Receptor preparation: selects A chain and cleans the structure from waters and heteroatoms 2.b) RDKit Molecules preparation: adds hydrogens and prepares the molecules for docking 3.a) Find pockets: uses P2Rank to predict the most promising ...
IsoAnnot
IsoAnnot is a new tool for generating functional and structural annotation at isoform level, capable of collecting and integrating information from different databases to categorize and describe each isoform, including functional and structural information for both transcript and protein.
⚠️⚠️ IsoAnnot is currenlty under beta-testing. Please see the latest release and download IsoAnnot from the ...
nf-synteny
A simple pipeline to run a macro synteny analysis.
It is under development, so if you wish to use the pipeline for your own research, please contact us (ecoflow.ucl [at] gmail.com). We can give you the up to date detail for the methods and the up to date figures. When it is published we will release a final version.
Synteny is the study of chromosome arrangement and gene order. Over evolutionary time, two species diverge from the state of the common ancestor, due to a variety of ...
Graphical and Interactive Spatial Proteomics Image Analysis Workflow
Open in Codespaces
Click the link above to open the workflow in GitHub Codespaces.
A demonstration video for this workflow is available at https://youtu.be/vHjdCAZQhfE
For more information on how to use Bwb, click this LINK.
Pre-print: ...
EGG: Epitope Generation Gateway
A modular Snakemake pipeline for personalized neoantigen discovery and prioritization using patient-specific gene co-expression networks.
![]()
Overview
EGG is a comprehensive computational workflow for identifying and prioritizing tumor neoantigens from DNA and RNA sequencing data. Unlike traditional neoantigen prediction pipelines that focus solely on ...
Type: Snakemake
Creators: Luca Mannino, Michele Fratello, Jacopo Chiaro, Federica D'Alessio, Emanuele Di Lieto, Miina Niittykoski, Angela Serra, Seppo Ylä-Herttuala, Vincenzo Cerullo, Antonio Federico, Dario Greco
Submitter: Luca Mannino

MPLID: Membrane Protein-Lipid Interface Dataset
![]()
![]()
![]()
![]()
A large-scale dataset of experimentally ...
Type: Python
Creators: Folorunsho Bright Omage, Ivan Mazoni, Inácio Henrique Yano, Goran Neshich
Submitter: Folorunsho Bright
MPLID: Membrane Protein-Lipid Interface Dataset
![]()
![]()
![]()
![]()
A large-scale dataset of experimentally ...
Type: Python
Creators: Folorunsho Bright Omage, Ivan Mazoni, Inácio Henrique Yano, Goran Neshich
Submitter: Folorunsho Bright
Segmentation and Reference Point Detection for Laser Capture Microdissection (LMD)
Project Summary
This repository contains the code for a Cellpose-SAM & pyLMD project dedicated to automating cell boundary and reference point detection in microscopic images used for Laser Capture Microdissection (LMD).
The primary function of this repository is to identify the boundaries of target cells and detect ...
NanoporeDB_workflow
![]()
1. Overview
This repository contains the integrated computational workflow for the large-scale mining, multimeric structure prediction, and quality filtering of protein nanopores. This pipeline enables the discovery of novel nanopore candidates from massive metagenomic and genomic databases. The structural models, pore geometry analysis, and ...
Automated image processing from movies to 2D classification. Includes quality and curator micrgographs protocols as Dose analysis, maxshift, tilt analysis, categorize micrographs, ctf consensus, also include particle curator as Remove duplicates and Deep micrograph cleaner. It also include a support branch to calculate the Box Size and train a model to pick the particles. The list of plugins required are: pwem, xmipp3, motioncorr, miffi, cistem, emfacilities, sphire, gautomatch, relion, repic
Identify glycan and polysaccharide compositions from mass spectrometry files. The workflow itself performs a conversion of raw files, GlyCombo search, and visualisation of results.
Example showing how to interact with the public bioimaging resources like the Image Data Resource (IDR)

Collection of EC-Earth3 workflows running with Autosubmit 3, with and without task wrappers, for different Slurm-based HPC platforms. See the individual descriptions for further details.
Collections of materials used as part of the Data Steward Curriculum
https://github.com/crim-ca/application-packages
Common Workflow Language definitions that can be employed in the context of OGC API - Processes deployment. Open Geospatial Consortium (OGC) API Processes can leverage CWL to integrate worklows and processes.
Open source Web API bridging OGC API - Processes with CWL by the Computer Research Institute of Montreal / Centre de recherche informatique de Montréal (CRIM): https://github.com/crim-ca/weaver
Collection of workflows exploring data in the Image Data Resource (IDR).

Open Work Flow Articles (OWFAs)
Maintainers: Mehmet Keçeci
Number of items: 25
Tags: Open Work flow Articles (OWFAs), Open Work flow Articles, OWFAs

This is an inclusive collection of workflows related to biodiversity and ecology (especially non-microbial). A big portion covers genome assembly of newly-sequenced species, using long reads (ONT or PacBio HiFi), possibly complemented by chromosome capture (typically HiC) for scaffolding, or/and by short reads (typically Illumina). It also aims at collating workflows related to ecology, biodiversity, biogeography, natural history, and related scientific areas, across the whole WorkflowHub and ...
Maintainers: Matúš Kalaš, Keiler Collier
Number of items: 28
Tags: Biodiversity, Ecology, Genome assembly, dna barcoding, natural history collections

A collection of image processing templates designed to support data collection and automated processing for Single Particle Analysis (SPA) in cryo-electron microscopy. The workflows range from simple pipelines—including movie alignment, CTF estimation, and quality assessment—to fully automated 2D and 3D processing. Please select the workflow that best suits your needs.
EuCanImage FHIR ETL Implementation
This repository contains the ETL implementation for EuCanImage, encouraging semantic interoperability of the clinical data obtained in the studies by transforming it into a machine-readable format following FHIR standards. This parser uses FHIR Resources in order to create the dictionaries following a FHIR compliant structure.
- Code Language is written in Python 3.11. ...

A set of generic and automatic workflows designed to:
-
Run on-the-fly and unattended.
-
Maintain robust stability for a wide range of samples.
-
Covers steps from movies to CTF estimation (for the moment).
-
Monitor the acquisition process and provide user feedback.
-
Comprise three proposed workflows, each with an additional layer of complexity.
TronFlow is an open source collection of computational workflows originally conceived for tumor-normal somatic variant calling over whole exome data and the manipulation of BAM and VCF files with the aim of having comparable and analysis-ready data. Over time, we have extended it to germline variant calling, copy numbers and other related technologies and analyses.
Its modular architecture covers different analytical and methodological use cases that allow analysing FASTQ files into analysis-ready ...
Maintainers: Pablo Riesgo Ferreiro
Number of items: 2
Tags: Nextflow, variant calling, VCF, Mutect2, HaplotyeCaller, Strelka2, Alignment, Annotation