plant2human workflow
 View on GitHub
View on GitHubplant2human workflow 🌾 ↔ 🕺

![]()

Introduction
This analysis workflow is centered on foldseek, which enables fast structural similarity searches and supports the discovery of understudied genes by comparing plants, which are distantly related species, with humans, for which there is a wealth of information. Based on the list of genes you are interested in, you can easily create a scatter plot of “structural similarity vs. sequence similarity” by retrieving structural data from the AlphaFold protein structure database (AFDB).
📣 Report
- ✔ 2025-12-13: main workflow update!
plant2human_v3_stringent.cwl(recommend) andplant2human_v3_permissive.cwl - ✔ 2026-01-21: Published in Bioinformatics Advances! The workflow version corresponding to the paper is available on WorkflowHub: main @ b1c1e73 (latest). We will keep updating it as the project evolves!
- ✔ 2026-03-22:
Workflow/plant2human_v3_stringent.cwlis update! - ✔ 2026-03-22: Update Oryza sativa 100 genes (Ensembl plants release 62) test example
- ✔ 2026-03-24: Update Arabidopsis thaliana 100 genes (Ensembl plants release 62) test example (details)
- ✔ 2025-03-24: Update Zea mays 100 genes (Ensembl plants release 62) test example (details)
- ✔ 2025-03-24: Update Solanum lycopersicum 100 genes (Ensembl plants release 62) test example (details)
- ✔ 2025-03-24: Update Glycine max 100 genes (Ensembl plants release 62) test example (details)
🔧 Implementation Background
In recent years, with the AlphaFold protein structure database, it has become easier to obtain protein structure prediction data and perform structural similarity searches even for plant species such as rice. Against this background, searching for hits with “low sequence similarity and high structural similarity” for the gene groups being focused on has become possible. This approach may allow us to discover proteins that are conserved in distantly related species and to consider the characteristics of these proteins based on the wealth of knowledge we have about humans.
📈 Analysis Environment
Prerequisites
- Docker / Orbstack
- cwltool >= 3.1.20250110105449
📝 Note: This workflow is based on Common Workflow Language (CWL). Please see the Official Document
⚠️ Prerequisites (Python Environment)
I've already checked python 3.11 and packages version below. Please install the following packages beforehand!
(Using Development Containers makes it easy to reproduce your execution environment!)
polars==1.39.2
matplotlib==3.10.8
seaborn==0.13.2
unipressed==1.4.0
papermill==2.7.0
Using Dev Containers (Docker and VScode extension)
Most processes, such as Foldseek, use container (BioContainers), but some involve processing with jupyter notebook, which requires the preparation of some python libraries (e.g., polars.). If you want to experiment with a simple workflow, you can create an analysis environment easily using Dev Containers system, a VScode extension. Using this environment, the version of the python library is probably the one used during development, so errors are unlikely to occur (see Dockerfile for the package version).
Please check the official website for Dev Container details.
The machine used for testing (2026-03-20)
- Machine: 🍎 MacBook Pro 🍎
- Chip: Apple M3 Max
- memory: 128 GB
🌾 Analysis Example ( Oryza sativa subsp.japonica 100 genes vs Homo sapiens) 🌾 (ver. 2026-03-20)
Here, we will explain how to use the list of 100 rice genes as an example.
0. Clone Repository
git clone https://github.com/yonesora56/plant2human.git
cd ./plant2human/
1. Creation of a TSV file of gene and UniProt ID correspondences 🧬
First, you need the following gene list tsv file.
📝 Note: Please set the column name as "From"
From
Os12g0269700
Os10g0410900
Os05g0403000
Os06g0127250
Os02g0249600
Os09g0349700
Os03g0735150
Os08g0547350
Os06g0282400
Os05g0576750
Os07g0216600
Os10g0164500
Os07g0201300
Os01g0567200
Os05g0563050
Os03g0660050
Os11g0436450
...
The following TSV file is required to execute the following workflow.
📝 Note: Network access required in this process!
From UniProt Accession
Os01g0104800 A0A0N7KC66
Os01g0104800 Q657Z6
Os01g0104800 Q658C6
Os01g0152300 Q9LGI2
Os01g0322300 A0A9K3Y6N1
Os01g0322300 Q657N1
Os01g0567200 A0A0N7KD66
Os01g0567200 Q657K0
Os01g0571133 A0A0P0V4A8
Os01g0664500 A0A8J8XFG3
Os01g0664500 Q5SN58
Os01g0810800 A0A8J8XDQ1
Os01g0810800 B7FAC9
Os01g0875300 A0A0P0VB72
Os01g0924300 A0A0P0VCB7
...
To do this, you need to follow the CWL workflow command below. This YAML file is the parameter file for the workflow, for example.
📁 Where to save: Place your YAML file in the job/ directory.
YAML Template for UniProt ID Mapping
Below is a template YAML file for the UniProt ID mapping process.
Copy this template and modify the parameters marked with # <-- CHANGE THIS!.
Example file: job/os_100genes_uniprot_idmapping.yml
# ---------- OUTPUT SETTINGS ----------
# Output notebook filename (string)
output_notebook_name: "your_species_uniprot_idmapping.ipynb" # <-- CHANGE THIS!
# ---------- INPUT FILE ----------
# Your gene list TSV file (column header must be "From")
gene_id_file:
class: File # <-- DO NOT CHANGE
format: edam:format_3475 # <-- DO NOT CHANGE
location: ./path/to/your_gene_list.tsv # <-- CHANGE THIS! (path to your gene list)
# ---------- UniProt API SETTINGS ----------
# For plant species, use "Ensembl_Genomes" as query database
uniprot_api_query_db: "Ensembl_Genomes" # <-- DO NOT CHANGE (for plants)
uniprot_api_target_db: "UniProtKB" # <-- DO NOT CHANGE
# ---------- OUTPUT DIRECTORY/FILE NAMES ----------
# Directory for AlphaFold info JSON files
json_dir_name: "your_species_afinfo_json" # <-- CHANGE THIS!
# Structure file format: "cifUrl" for mmCIF file format (recommended), "pdbUrl" for PDB file format
data_url: "cifUrl" # <-- Usually DO NOT CHANGE
# Directory for downloaded structure files
structure_dir_name: "your_species_mmcif" # <-- CHANGE THIS!
# Output TSV filename for ID mapping results
id_mapping_all_file_name: "your_species_idmapping_all.tsv" # <-- CHANGE THIS!
Command Execution Example
# test date: 2026-03-20
cwltool --debug --outdir ./test/oryza_sativa_test_100genes_202603/ \
./Tools/01_uniprot_idmapping.cwl \
./job/os_100genes_uniprot_idmapping.yml
In this execution, mmcif files are also retrieved from AlphaFold Database (version 6). The execution results are output with the jupyter notebook format.
2. Creating and Preparing Indexes 📂
I'm sorry, but the main workflow does not currently include the creation of an index process (both for protein structure (foldseek index) and protein sequence (BLAST index)). Please perform the following processes in advance.
⚠️ Important: Database Version Compatibility ⚠️
This workflow uses data from the AlphaFold Protein Structure Database (AFDB) version 6. Due to recent database updates (v4 → v6, October 2025), users should be aware of potential version mismatches between different data components.
Understanding the Version Issue
| Component | AFDB Version | Source |
|---|---|---|
| Query structures (your plant proteins) | v6 | AlphaFold Database API |
| ⚠️ Foldseek pre-built index | v4 | foldseek databases command |
| ⭐ Foldseek index (built myself) | v6 | FTP download from AFDB |
sequences.fasta (for BLAST index) |
v6 | FTP download from AFDB |
Two Main Workflow Options
We provide two workflow variants to address this version compatibility issue:
| Workflow | Index Source | AFDB index Version Match | Database Options | Use Case |
|---|---|---|---|---|
plant2human_v3_permissive.cwl |
foldseek databases (pre-built) |
❌ v4 vs v6 | UniProt50, Swiss-Prot, Proteome, etc. | Exploratory analysis (swiss-prot,TrEMBL) |
plant2human_v3_stringent.cwl |
foldseek createdb (self-built) |
✅ v6 = v6 | Human proteome only | Rigorous analysis |
Option 1: Permissive Mode
Pros:
- Easy setup with
foldseek databasescommand - Access to diverse databases (UniProt50, Swiss-Prot, etc.)
Cons:
- Version mismatch between query (v6) and index (v4)
- Some proteins may have updated structures in v6 that differ from v4
When to use: broad searches (including swiss-prot, TrEMBL)
➡️ Go to: 2-1a. Creating a Foldseek Index (Option 1: Permissive Mode))
Option 2: Stringent Mode (Recommended)
Pros:
- Full version consistency (v6 query ↔ v6 index)
- Smaller index size (Human proteome only: ~24,000 proteins)
- Reproducible results with matched database versions
Cons:
- Requires manual download and index creation
- Limited to Human proteome only
When to use: Final analysis for publications, when version consistency is critical
➡️ Go to: 2-1b. Creating Index (Stringent Mode)
2-1. Creating a Foldseek Index for structural alignment
2-1a. Creating a Foldseek Index (Option 1: Permissive Mode)
In this workflow, the target of the structural similarity search is specified as the AlphaFold database to perform comparisons across a broader range of species.
Index creation using the foldseek databases command is through the following command.
Please select the database you want to use from Alphafold/UniProt, Alphafold/UniProt50-minimal, Alphafold/UniProt50, Alphafold/Proteome, Alphafold/Swiss-Prot.
# Supported databases in this workflow
Alphafold/UniProt
Alphafold/UniProt50-minimal
Alphafold/UniProt50
Alphafold/Proteome
Alphafold/Swiss-Prot
You can check the details of this database using the following command.
docker run --rm quay.io/biocontainers/foldseek:10.941cd33--h5021889_1 foldseek databases --help
For example, if you want to specify AlphaFold/Swiss-Prot as the index, you can do so with the following CWL file;
# execute creation of foldseek index using "foldseek databases"
# test date: 2025-12-12
cwltool --debug --outdir ./index/ \
./Tools/02_foldseek_database.cwl \
--database Alphafold/Swiss-Prot \
--index_dir_name index_swissprot \
--index_name swissprot \
--threads 16
2-1b. Creating a Foldseek Index (Option 2: Stringent Mode)
In this mode, you download structure files directly from AFDB v6 and create your own index. This ensures version consistency between query and target structures.
Step 1: Download Human proteome from AFDB v6
# Download date: 2026-03-20
# file size is ~5GB
cd ./index
# curl
curl -O https://ftp.ebi.ac.uk/pub/databases/alphafold/v6/UP000005640_9606_HUMAN_v6.tar
# or aria2c
aria2c -c --max-connection-per-server=4 \
--min-split-size=1M \
-o "UP000005640_9606_HUMAN_v6.tar" \
"https://ftp.ebi.ac.uk/pub/databases/alphafold/v6/UP000005640_9606_HUMAN_v6.tar"
cd ../
Step 2: Create Foldseek index using foldseek createdb command
# test date: 2026-03-20
# foldseek version: https://github.com/steineggerlab/foldseek/releases/tag/10-941cd33
cwltool --debug --outdir ./index/ \
./Tools/02_foldseek_createdb.cwl \
--input_structure_files ./index/UP000005640_9606_HUMAN_v6.tar \
--index_dir_name index_human_proteome_v6 \
--index_name human_proteome_v6 \
--threads 16
2-2. Creating a Index for protein "sequence" alignment (Common)
An index protein sequence FASTA file must be downloaded to obtain the amino acid sequence using the blastdbcmd command from the AlphaFold Protein Structure Database.
This workflow uses the version of the protein sequence that was used for structure prediction.
📝 Note:: This FASTA file is extremely large (> 109GB !), so it's probably best to delete FASTA file after creating the index.
# Preparation for BLAST index
# test date: 2026-03-21
cd ./index
# curl
curl -O https://ftp.ebi.ac.uk/pub/databases/alphafold/sequences.fasta # AFDB version 6
# or aria2c (recommend)
aria2c --continue=true \
--max-connection-per-server=4 \
--min-split-size=1M \
https://ftp.ebi.ac.uk/pub/databases/alphafold/sequences.fasta # AFDB version 6
# rename
mv sequences.fasta afdb_all_sequences_v6.fasta
cd ../
# execute creation of BLAST index using "makeblastdb"
# test date: 2026-03-21
cwltool --debug \
--outdir ./index/ \
./Tools/03_makeblastdb.cwl \
--index_dir_name index_uniprot_afdb_all_sequences_v6 \
--input_fasta_file ./index/afdb_all_sequences_v6.fasta
📝 Note: It is estimated to take 2~ hours for creating index. This index is about > 150GB! We are currently investigating whether it can be executed by another method...
3. Execution of the plant2human workflow (main workflow)
📝 Note: Network access required in this process!
In this process, we perform a structural similarity search using the foldseek easy-search command and then perform a pairwise sequence alignment of the amino acid sequences of the hit pairs using the needle and water commands.
Finally, based on this information, we create a scatter plot and output a jupyter notebook as a report.
📝 Note: For Permissive Mode (using pre-built indexes like Swiss-Prot), see Workflow/README.md.
📋 YAML Parameter File Reference (Stringent Mode)
The main workflow requires a YAML parameter file to specify input files and parameters. Below is a detailed explanation of each parameter.
Example file (2026-03-22 update!): job/plant2human_v3_stringent_example_os100.yml
Input File Parameters
| Parameter | Type | Description | Example |
|---|---|---|---|
INPUT_DIRECTORY |
Directory | Directory containing mmCIF structure files from Step 1 | ../test/.../os_100_genes_mmcif/ |
FILE_MATCH_PATTERN |
string | File pattern for structure files | "*.cif" |
FOLDSEEK_INDEX |
File | Foldseek index created in Step 2-1b | ../index/index_human_proteome_v6/human_proteome_v6 |
QUERY_IDMAPPING_TSV |
File | ID mapping TSV from Step 1 | ..._idmapping_all.tsv |
QUERY_GENE_LIST_TSV |
File | Original gene list TSV | oryza_sativa_random_100genes_list.tsv |
Foldseek Parameters (foldseek easy-search command)
for more details, please execute the below command.
docker run --rm quay.io/biocontainers/foldseek:10.941cd33--h5021889_1 \
foldseek easy-search --help
| Parameter | Default | Description |
|---|---|---|
COVERAGE_THRESHOLD |
0.75 |
(0~1) Coverage threshold for search results |
COV_MODE |
5 |
(1,2,3,4,5) Coverage mode for search results. 5 means short sequence needs to be at least x% of the other seq. length |
EVALUE |
0.1 |
E-value threshold for structural similarity search |
ALIGNMENT_TYPE |
2 |
0: 3Di only, 1: TM-align (default), 2: 3Di+AA |
THREADS |
16 |
Number of CPU threads |
SPLIT_MEMORY_LIMIT |
"120G" |
Memory limit for large searches |
YAML Template for Stringent Mode
Copy and modify this template for your analysis:
# ============================================================
# YAML Parameter File for plant2human_v3_stringent.cwl
# Species: [Your Species Name]
# ============================================================
# ---------- INPUT DIRECTORY ----------
INPUT_DIRECTORY:
class: Directory
location: ./path/to/your_mmcif_directory/ # <-- CHANGE THIS!
FILE_MATCH_PATTERN: "*.cif"
# ---------- FOLDSEEK INDEX (Stringent Mode) ----------
FOLDSEEK_INDEX:
class: File
location: ../index/index_human_proteome_v6/human_proteome_v6 # <-- Adjust path if needed
secondaryFiles: # <-- If you do not place the index in the “index” directory, you must specify the path to all generated index files! (This is generally not required.)
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_ca
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_ca.dbtype
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_ca.index
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_h
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_h.dbtype
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_h.index
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_mapping
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_ss
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_ss.dbtype
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6_ss.index
# No _taxonomy for self-built index
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6.dbtype
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6.index
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6.lookup
- class: File
location: ../index/index_human_proteome_v6/human_proteome_v6.source
# No .version for self-built index
# ---------- FOLDSEEK DEFAULT PARAMETERS ----------
COVERAGE_THRESHOLD: 0.75
COV_MODE: 5
EVALUE: 0.1
ALIGNMENT_TYPE: 1 # 1 = TM-align
THREADS: 16
SPLIT_MEMORY_LIMIT: "120G"
# ---------- EXTRACT ID COLUMNS ----------
WF_COLUMN_NUMBER_QUERY_SPECIES: 1
WF_COLUMN_NUMBER_HIT_SPECIES: 2
QUERY_IDMAPPING_TSV:
class: File
format: edam:format_3475
location: ./path/to/your_idmapping_all.tsv # <-- CHANGE THIS!
QUERY_GENE_LIST_TSV:
class: File
format: edam:format_3475
location: ./path/to/your_gene_list.tsv # <-- CHANGE THIS!
Command Execution Example (Stringent Mode - Recommended)
# test date: 2026-03-22
cwltool --debug --outdir ./test/oryza_sativa_test_100genes_202603/ \
./Workflow/plant2human_v3_stringent.cwl \
./job/plant2human_v3_stringent_example_os100.yml
The execution results are output with the jupyter notebook.
📝 Note: For more detailed analysis or to modify the parameters in the figure, you can interactively operate this notebook again yourself! (2026-03-22) We have configured it to output the TSV files and scatter plot images generated in the Jupyter notebook! All result is generated as TSV file
📝 Note: For Permissive Mode (using pre-built indexes like Swiss-Prot), see Workflow/README.md.
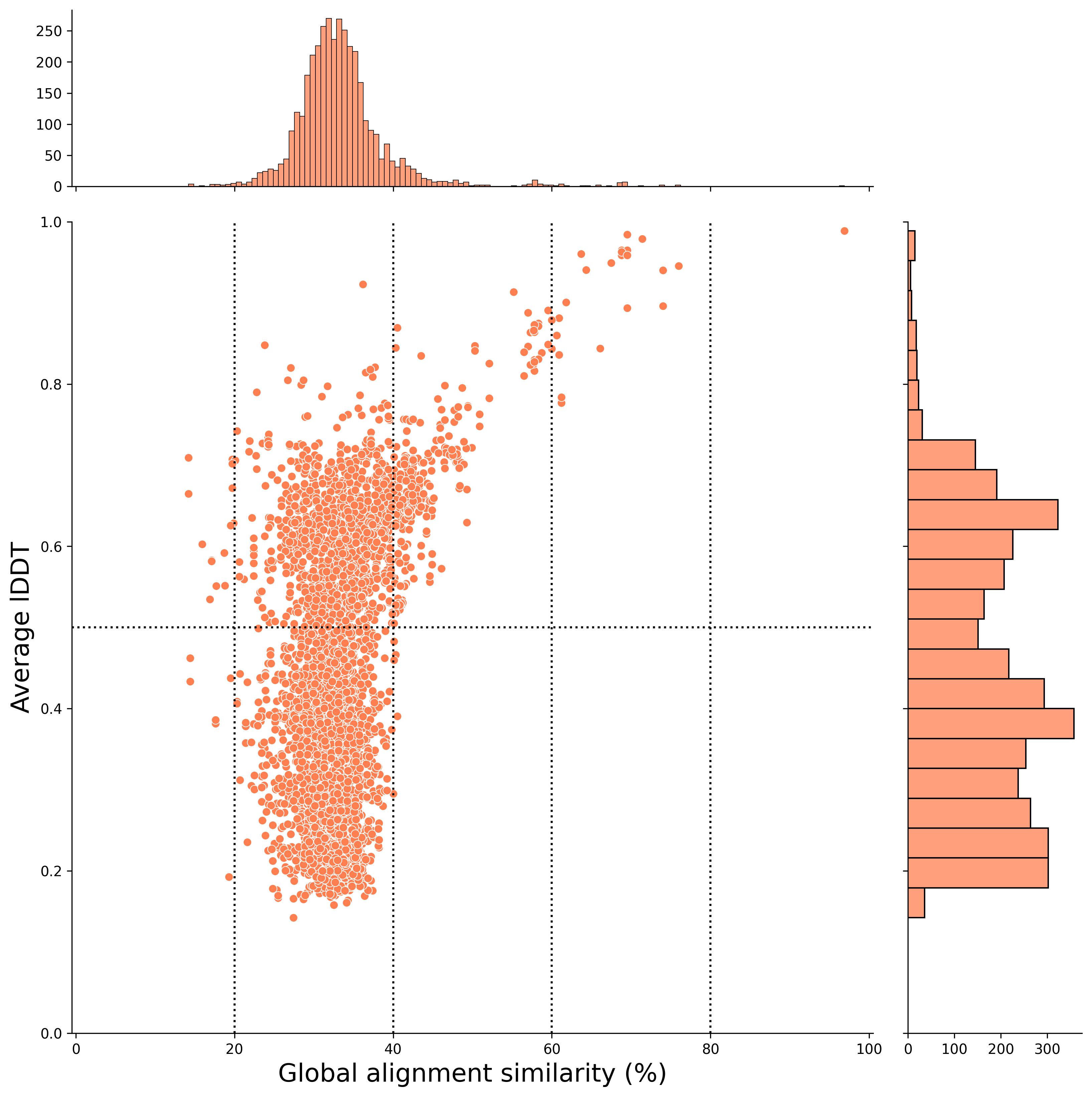
rice vs human result (strngent mode result) 🌾 ↔ 🕺
For example, you can visualize the results of structural similarity and global alignment, as shown below. In this case, the x-axis represents the global alignment similarity match (%), and the y-axis represents the average lDDT score (an indicator of structural alignment).
The hit pairs in the upper-right plot indicate higher sequence similarity and structural similarity.
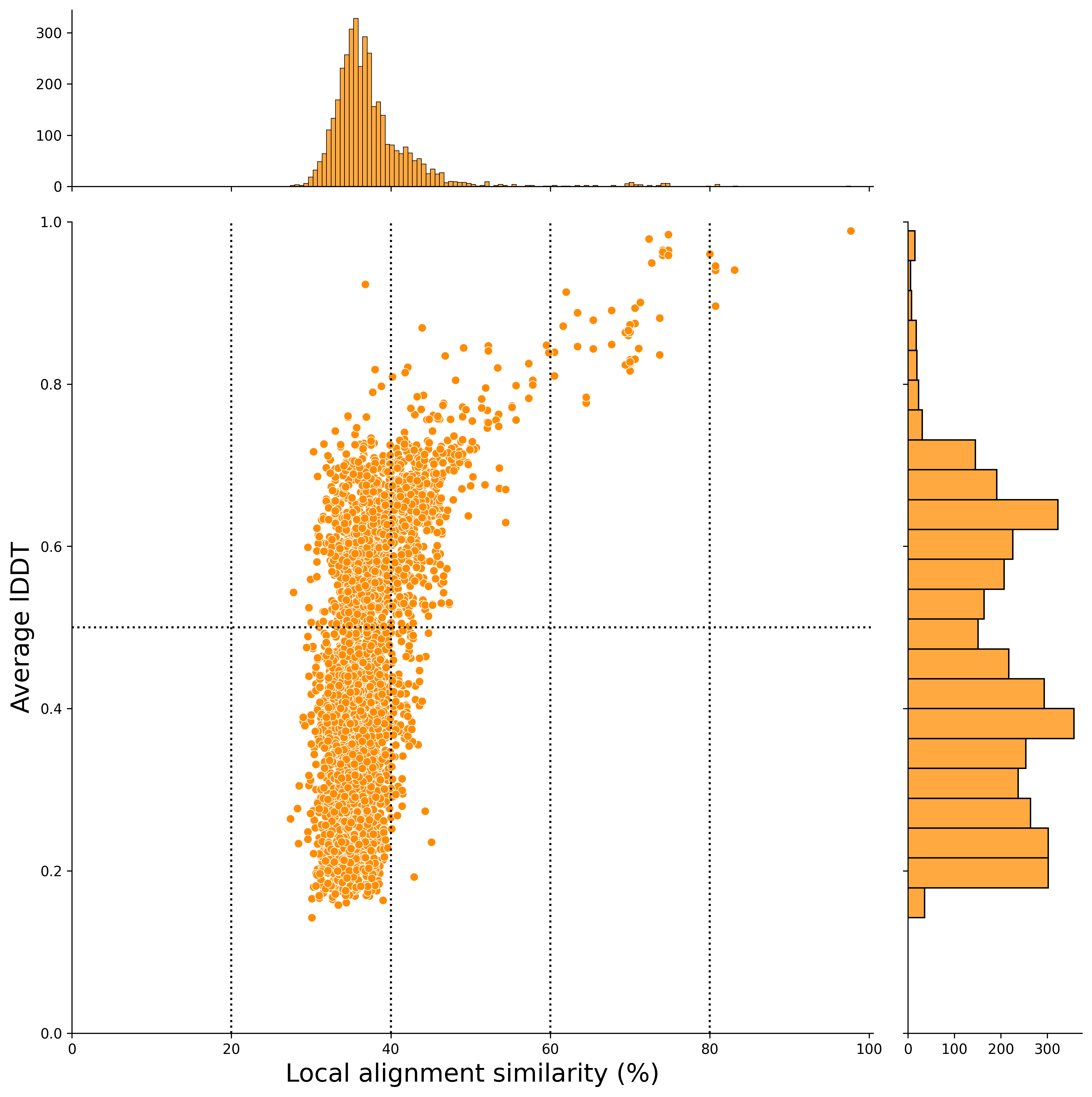
In this case, the x-axis represents the local alignment similarity match (%), and the y-axis represents the average lDDT score (an indicator of structural alignment).
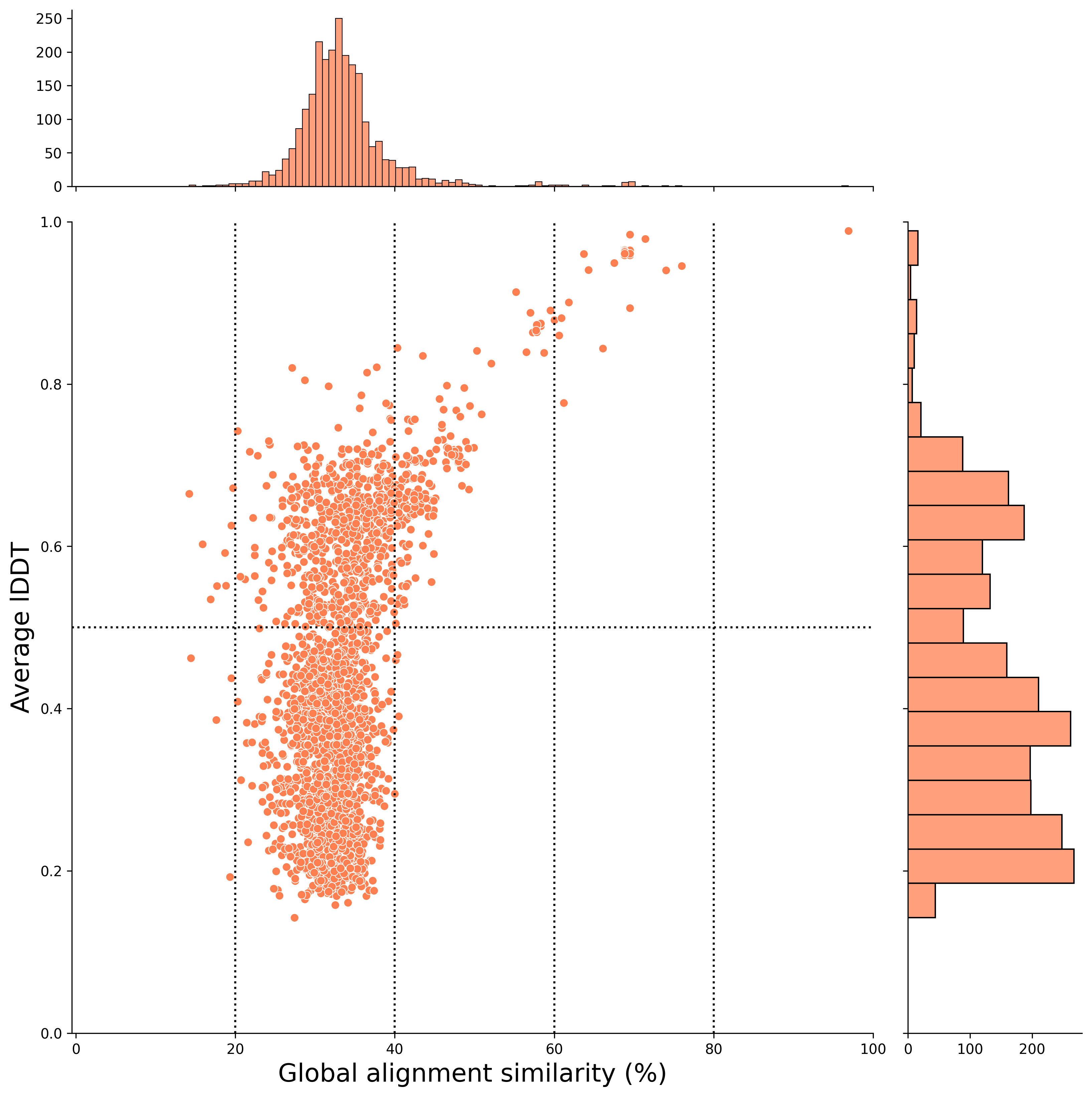
After Filtering
The report notebook for the plant2human workflow also outputs scatter plots after applying the filtering conditions set in this workflow.
Filtering criteria
- structural alignment coverage >= 50%
- If there are hits with the same target for the same gene-derived UniProt ID, the one with the highest qcov is selected, and if the qcov is the same, the one with the highest lDDT is selected.
📝 Note: In this workflow, we leave the states with the same foldseek hit even if the query genes are different.
- Select hits that can be converted to Ensembl gene id and HGNC Gene nomenclature with TogoID API
By applying these filtering conditions, you can examine hit pairs that are easier to investigate!
Global alignment (x-axis) (After Filtering)
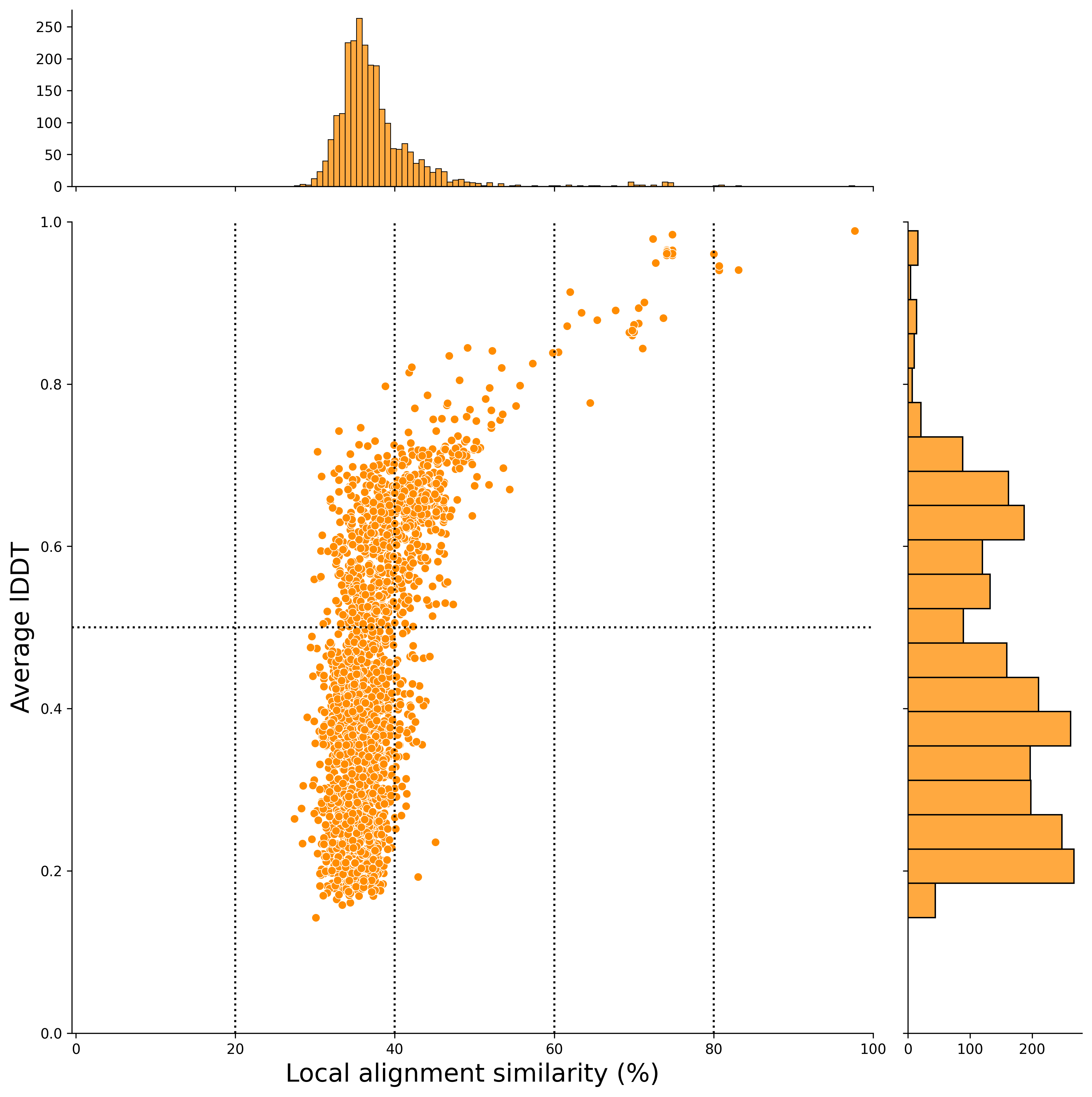
local alignment (x-axis) (After Filtering)
🌿 Running the Pipeline for Another Plant Species 🌿
This workflow can be applied to any plant species available in the AlphaFold Protein Structure Database (AFDB).
Step 1: Check Species Availability in AFDB
Before running the pipeline, verify that your target plant species is available:
- Visit AFDB Page
- Search for your species name or UniProt proteome ID
- Confirm protein structures are available for your genes
📝 Note: Most model organisms and many crop species are available in AFDB v6.
Step 2: Prepare Your Gene List
Create a TSV file with column header "From" containing your gene IDs:
| Species | Gene ID Format | Example |
|---|---|---|
| Oryza sativa | RAP-DB format | Os01g0104800 |
| Arabidopsis thaliana | TAIR format | AT1G01010 |
| Zea mays | Ensembl format | Zm00001eb000010 |
| Solanum lycopersicum | Ensembl format | Solyc01g005000 |
| Glycine max | Ensembl format | Glyma.01G000100 |
Step 3: Create YAML Parameter Files
- Copy the template from Section 1 (UniProt ID Mapping) and Section 3 (Main Workflow)
- Modify the paths and filenames marked with
# <-- CHANGE THIS! - Save your YAML files in the
job/directory
Step 4: Execute the Workflow
Follow the same steps as described in Sections 1-3, using your custom YAML files.
📚 Example Implementations for Other Species
We provide complete examples for multiple plant species. Use these as references:
| Species | Test Directory | YAML Files |
|---|---|---|
| Arabidopsis thaliana | test/arabidopsis_test_100genes_202603/ |
job/at_100genes_*.yml |
| Zea mays | test/zea_mays_test_100genes_202603/ |
job/zm_100genes_*.yml |
| Solanum lycopersicum | test/solanum_lycopersicum_test_100genes_202603/ |
job/sl_100genes_*.yml |
| Glycine max | test/glycine_max_test_100genes_202603/ |
job/gm_100genes_*.yml |
Click and drag the diagram to pan, double click or use the controls to zoom.
Inputs
| ID | Name | Description | Type |
|---|---|---|---|
| INPUT_DIRECTORY | input protein structure files directory | query protein structure file (default: mmCIF) directory for foldseek easy-search input. |
|
| FILE_MATCH_PATTERN | structure file match pattern | file match pattern for listing input files. default: *.cif |
|
| FOLDSEEK_INDEX | foldseek index files | "foldseek index files for foldseek easy-search input. default: ../index/index_swissprot/swissprot Note: At this time (2025/02/02), the process of acquiring and indexing index files for execution has not been incorporated into the workflow. Therefore, we would like you to execute the following commands in advance. example: `foldseek databases Alphafold/Swiss-Prot index_swissprot/swissprot tmp --threads 8` " |
|
| COVERAGE_THRESHOLD | coverage threshold (foldseek easy-search) | coverage threshold for foldseek easy-search. default: 0.75 |
|
| COV_MODE | coverage mode (foldseek easy-search) | coverage mode for foldseek easy-search. for more details, see `foldseek easy-search --help` |
|
| EVALUE | e-value (foldseek easy-search) | e-value threshold for foldseek easy-search. workflowdefault: 0.1 |
|
| ALIGNMENT_TYPE | alignment type (foldseek easy-search) | alignment type for foldseek easy-search. default: 1 (TM-align) for detailed information, see foldseek GitHub repository. |
|
| THREADS | threads (foldseek easy-search) | threads for foldseek easy-search. default: 16 |
|
| SPLIT_MEMORY_LIMIT | split memory limit (foldseek easy-search) | split memory limit for foldseek easy-search. default: 120G |
|
| WF_COLUMN_NUMBER_QUERY_SPECIES | column number of query species | column number of query species. default: 1 (UniProt ID list) |
|
| OUTPUT_FILE_NAME_QUERY_SPECIES | output file name (extract query species column) | output file name for extract query species column python script. default: foldseek_result_query_species.txt |
|
| WF_COLUMN_NUMBER_HIT_SPECIES | column number of hit species | column number of hit species. default: 2 (UniProt ID list) |
|
| OUTPUT_FILE_NAME_HIT_SPECIES | output file name (extract hit species column) | output file name for extract hit species column python script. default: foldseek_result_hit_species.txt |
|
| BLAST_INDEX_FILES | blast index files (blastdbcmd) | blast index files for blastdbcmd |
|
| QUERY_IDMAPPING_TSV | query idmapping tsv (papermill process) | query idmapping tsv file. Retrieve files in advance. default: rice UniProt ID mapping file |
|
| QUERY_GENE_LIST_TSV | query gene list tsv (papermill process) | query gene list tsv file. Retrieve files in advance. default: rice random gene list |
|
| FOLDSEEK_RESULT_PARSE_NOTEBOOK | jupyter notebook for parse workflow results | jupyter notebook template for parsing workflow results (Stringent Mode) |
|
Steps
| ID | Name | Description |
|---|---|---|
| sub_workflow_foldseek_easy_search | foldseek easy-search sub-workflow process | "Execute foldseek easy-search using foldseek using BioContainers docker image. This workflow supports only TSV file output. Step 1: listing files Step 2: foldseek easy-search process" |
| extract_target_species | extract target species (human) process | Extract target species (human) from foldseek easy-search result. execute: ../Tools/12_extract_target_species.cwl |
| extract_query_species_column | extract query species column process | Extract query species column (UniProt ID list) from foldseek easy-search result. execute: ../Tools/13_extract_id.cwl |
| extract_hit_species_column | extract hit species column process | Extract hit species column (UniProt ID list) from foldseek easy-search result. execute: ../Tools/13_extract_id.cwl |
| sub_workflow_retrieve_sequence_query_species | retrieve sequence sub-workflow process using EMBOSS package | "Perform pairwise alignment of protein sequences for pairs identified by structural similarity search. Step 1: blastdbcmd: ../Tools/14_blastdbcmd.cwl Step 2: seqretsplit: ../Tools/15_seqretsplit.cwl Step 3: needle (Global alignment): ../Tools/16_needle.cwl Step 4: water (Local alignment): ../Tools/16_water.cwl " |
| togoid_convert | togoid convert process | retrieve UniProt ID to HGNC gene symbol using togoID python script. execute: ../Tools/17_togoid_convert.cwl |
| papermill | papermill process | output notebook using papermill. This process allows you to create a scatter plot of structural similarity vs. sequence similarity. execute: ../Tools/18_papermill.cwl |
Outputs
| ID | Name | Description | Type |
|---|---|---|---|
| IDLIST1 | output file (extract query species column) | extract query species column UniProt ID list file. |
|
| IDLIST2 | output file (extract hit species column) | extract hit species column UniProt ID list file. |
|
| BLASTDBCMD_RESULT1 | blastdbcmd result (query species) | blastdbcmd result file for query species. |
|
| BLASTDBCMD_RESULT2 | blastdbcmd result (hit species) | blastdbcmd result file for hit species. |
|
| LOGFILE1 | logfile (blastdbcmd query species) | logfile for blastdbcmd query species. |
|
| LOGFILE2 | logfile (blastdbcmd hit species) | logfile for blastdbcmd hit species. |
|
| DIR1 | directory (seqretsplit query species) | directory for seqretsplit query species. |
|
| FASTA_FILES1 | split fasta files (seqretsplit query species) | split fasta files using seqretsplit for pairwise sequence alignment. |
|
| DIR2 | directory (seqretsplit hit species) | directory for seqretsplit hit species. |
|
| FASTA_FILES2 | split fasta files (seqretsplit hit species) | split fasta files using seqretsplit for pairwise sequence alignment. |
|
| DIR3 | needle result directory | needle (global alignment) result directory. |
|
| NEEDLE_RESULT_FILE | needle result file (.needle) | needle (global alignment) result files. suffix is .needle. |
|
| DIR4 | water result directory | water (local alignment) result directory. |
|
| WATER_RESULT_FILE | water result file (.water) | water (local alignment) result files. suffix is .water. |
|
| TSVFILE3 | output file (togoid convert) | output file for togoid convert. |
|
| REPORT_NOTEBOOK | output notebook (papermill) | output notebook using papermill. notebook name is `plant2human_report.ipynb`. |
|
| FOLDSEEK_RESULT_JOIN_ALIGNMENT_RESULT_ALL | foldseek result join alignment result all | foldseek result join alignment result all |
|
| FOLDSEEK_RESULT_JOIN_ALIGNMENT_RESULT_FILTER | foldseek result join alignment result filter | foldseek result join alignment result filter |
|
| FOLDSEEK_RESULT_GENE_LEVEL_HIT_COUNT_ALL | foldseek result gene level hit count all | foldseek result gene level hit count all |
|
| FOLDSEEK_RESULT_SCATTER_PLOT | foldseek result scatter plot | foldseek result scatter plot |
|
Version History
main @ 62a2b67 (latest) Created 24th Mar 2026 at 02:32 by Sora Yonezawa
plant2human_stringent_v3.cwl is updated!
Frozen
 main
main62a2b67
main @ b1c1e73 Created 18th Dec 2025 at 01:36 by Sora Yonezawa
This workflow version corresponds to the article! Please see: https://doi.org/10.1093/bioadv/vbag013
Frozen
main
b1c1e73
main @ fc8edcd Created 13th Dec 2025 at 10:03 by Sora Yonezawa
add tomato and soybean example
Frozen
main
fc8edcd
main @ 10d8268 Created 3rd Sep 2025 at 21:20 by Sora Yonezawa
update oryza sativa 100genes test
Frozen
main
10d8268
main @ 6911e7a Created 13th Jul 2025 at 05:00 by Sora Yonezawa
update foldseek database process
Frozen
main
6911e7a
main @ 76a6471 Created 20th Nov 2024 at 05:29 by Sora Yonezawa
update README & add description
Frozen
main
76a6471
main @ 1aa2763 Created 16th Nov 2024 at 10:34 by Sora Yonezawa
main workflow changed
Frozen
main
1aa2763
main @ b8c0b1d (earliest) Created 16th Nov 2024 at 04:56 by Sora Yonezawa
UPDATE README & zey mays test files
Frozen
main
b8c0b1d
 Creators and Submitter
Creators and Submitter
Views: 14061 Downloads: 3336
Created: 16th Nov 2024 at 04:56
Last updated: 24th Mar 2026 at 02:35
AttributionsNone
Related items

 https://orcid.org/0009-0004-1874-3117
https://orcid.org/0009-0004-1874-3117Hiroshima University, Graduate School of Integrated Sciences for life, Laboratory of Genome Informatics, Ph.D student GitHub: https://github.com/yonesora56

Toward data-driven genome breeding (digital breeding), we are developing data analysis infrastructure technology essential for genome editing, focusing on gene function analysis using bioinformatics called BioDX.
Space: Hiroshima workflow community
Public web page: https://bonohu.hiroshima-u.ac.jp/index_en.html