VariantCaller_GATK3.6 Version 1
 View on GitHub
View on GitHubRare disease researchers workflow is that they submit their raw data (fastq), run the mapping and variant calling RD-Connect pipeline and obtain unannotated gvcf files to further submit to the RD-Connect GPAP or analyse on their own.
This demonstrator focuses on the variant calling pipeline. The raw genomic data is processed using the RD-Connect pipeline (Laurie et al., 2016) running on the standards (GA4GH) compliant, interoperable container orchestration platform.
This demonstrator will be aligned with the current implementation study on Development of Architecture for Software Containers at ELIXIR and its use by EXCELERATE use-case communities
For this implementation, different steps are required:
- Adapt the pipeline to CWL and dockerise elements
- Align with IS efforts on software containers to package the different components (Nextflow)
- Submit trio of Illumina NA12878 Platinum Genome or Exome to the GA4GH platform cloud (by Aspera or ftp server)
- Run the RD-Connect pipeline on the container platform
- Return corresponding gvcf files
- OPTIONAL: annotate and update to RD-Connect playground instance
N.B: The demonstrator might have some manual steps, which will not be in production.
RD-Connect pipeline
Detailed information about the RD-Connect pipeline can be found in Laurie et al., 2016
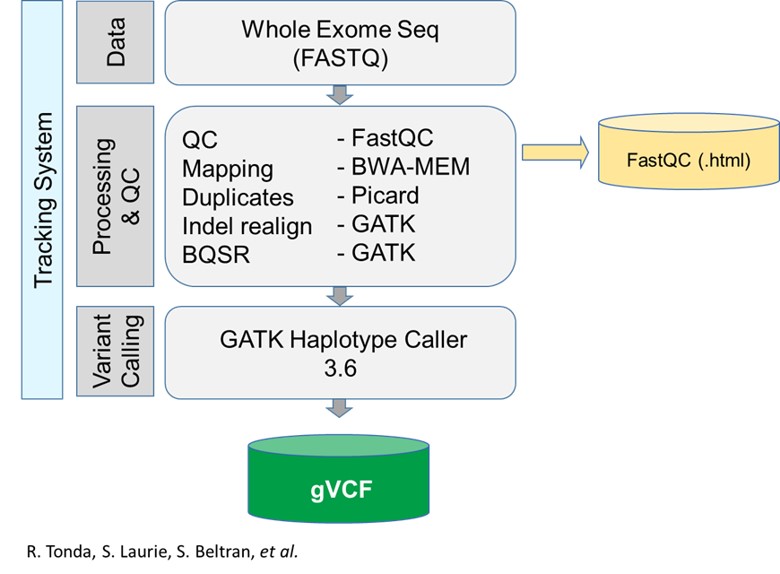
The applications
1. Name of the application: Adaptor removal
Function: remove sequencing adaptors
Container (readiness status, location, version): cutadapt (v.1.18)
Required resources in cores and RAM: current container size 169MB
Input data (amount, format, directory..): raw fastq
Output data: paired fastq without adaptors
2. Name of the application: Mapping and bam sorting
Function: align data to reference genome
Container : bwa-mem (v.0.7.17) / Sambamba (v. 0.6.8 )(or samtools)
Resources :current container size 111MB / 32MB
Input data: paired fastq without adaptors
Output data: sorted bam
3. Name of the application: MarkDuplicates
Function: Mark (and remove) duplicates
Container: Picard (v.2.18.25)
Resources: current container size 261MB
Input data:sorted bam
Output data: Sorted bam with marked (or removed) duplicates
4. Name of the application: Base quality recalibration (BQSR)
Function: Base quality recalibration
Container: GATK (v.3.6-0)
Resources: current container size 270MB
Input data: Sorted bam with marked (or removed) duplicates
Output data: Sorted bam with marked duplicates & base quality recalculated
5. Name of the application: Variant calling
Function: variant calling
Container: GATK (v.3.6-0)
Resources: current container size 270MB
Input data:Sorted bam with marked duplicates & base quality recalculated
Output data: unannotated gvcf per sample
6. (OPTIONAL)Name of the application: Quality of the fastq
Function: report on the sequencing quality
Container: fastqc 0.11.8
Resources: current container size 173MB
Input data: raw fastq
Output data: QC report
Licensing
GATK declares that archived packages are made available for free to academic researchers under a limited license for non-commercial use. If you need to use one of these packages for commercial use. https://software.broadinstitute.org/gatk/download/archive
Click and drag the diagram to pan, double click or use the controls to zoom.
Inputs
| ID | Name | Description | Type |
|---|---|---|---|
| fastq_files | n/a | List of paired-end input FASTQ files |
|
| reference_genome | n/a | Compress FASTA files with the reference genome chromosomes |
|
| known_indels_file | n/a | VCF file correlated to reference genome assembly with known indels |
|
| known_sites_file | n/a | VCF file correlated to reference genome assembly with know sites (for instance dbSNP) |
|
| chromosome | n/a | Label of the chromosome to be used for the analysis. By default all the chromosomes are used |
|
| readgroup_str | n/a | Parsing header which should correlate to FASTQ files |
|
| sample_name | n/a | Sample name |
|
| gqb | n/a | Exclusive upper bounds for reference confidence GQ bands (must be in [1, 100] and specified in increasing order) |
|
Steps
| ID | Name | Description |
|---|---|---|
| unzipped_known_sites | n/a | n/a |
| unzipped_known_indels | n/a | n/a |
| gunzip | n/a | n/a |
| picard_dictionary | n/a | n/a |
| cutadapt2 | n/a | n/a |
| bwa_index | n/a | n/a |
| samtools_index | n/a | n/a |
| bwa_mem | n/a | n/a |
| samtools_sort | n/a | n/a |
| picard_markduplicates | picard-MD | n/a |
| gatk3-rtc | gatk3-rtc | n/a |
| gatk-ir | gatk-ir | n/a |
| gatk-base_recalibration | gatk-base_recalibration | n/a |
| gatk-base_recalibration_print_reads | gatk-base_recalibration_print_reads | n/a |
| gatk_haplotype_caller | gatk-haplotype_caller | n/a |
Outputs
| ID | Name | Description | Type |
|---|---|---|---|
| metrics | n/a | Several metrics about the result |
|
| gvcf | n/a | unannotated gVCF output file from the mapping and variant calling pipeline |
|
Version History
Version 1 (earliest) Created 18th Feb 2021 at 15:01 by Laura Rodriguez-Navas
Added/updated 1 files
Open
 master
mastere0f868f
 Creators and Submitter
Creators and Submitter

Views: 5637 Downloads: 1247
Created: 18th Feb 2021 at 15:01
Last updated: 8th Mar 2021 at 21:35
Tags AttributionsNone
Related items


Teams: GalaxyProject SARS-CoV-2, nf-core viralrecon, EOSC-Life - Demonstrator 7: Rare Diseases, iPC: individualizedPaediatricCure, EJPRD WP13 case-studies workflows, TransBioNet, OpenEBench, ELIXIR Proteomics
Organizations: Barcelona Supercomputing Center, ELIXIR
 https://orcid.org/0000-0003-4929-1219
https://orcid.org/0000-0003-4929-1219
Computer Engineer in Barcelona Supercomputing Center (BSC)

EOSC-Life brings together the 13 Life Science ‘ESFRI’ research infrastructures (LS RIs) to create an open, digital and collaborative space for biological and medical research.
The project will publish ‘FAIR’ data and a catalogue of services provided by participating RIs for the management, storage and reuse of data in the European Open Science Cloud (EOSC).
Teams: EOSC-Life - Demonstrator 7: Rare Diseases, EOSC-Life WP3
Web page: https://www.eosc-life.eu/

An integrative analysis pipeline of genomic and transcriptomic human data for disentangling the genetic origin of a rare-disease in the context of the European Open Science Cloud.
Space: EOSC-Life
Public web page: https://www.eosc-life.eu/services/demonstrators/