Jupyter Notebook Mutation Free Energy Calculations
 View on GitHub
View on GitHubMutation Free Energy Calculations using BioExcel Building Blocks (biobb)
Based on the official pmx tutorial.
This tutorial aims to illustrate how to compute a fast-growth mutation free energy calculation, step by step, using the BioExcel Building Blocks library (biobb). The particular example used is the Staphylococcal nuclease protein (PDB code 1STN), a small, minimal protein, appropriate for a short tutorial.
The non-equilibrium free energy calculation protocol performs a fast alchemical transition in the direction WT->Mut and back Mut->WT. The two equilibrium trajectories needed for the tutorial, one for Wild Type (WT) and another for the Mutated (Mut) protein (Isoleucine 10 to Alanine -I10A-), have already been generated and are included in this example. We will name WT as stateA and Mut as stateB.
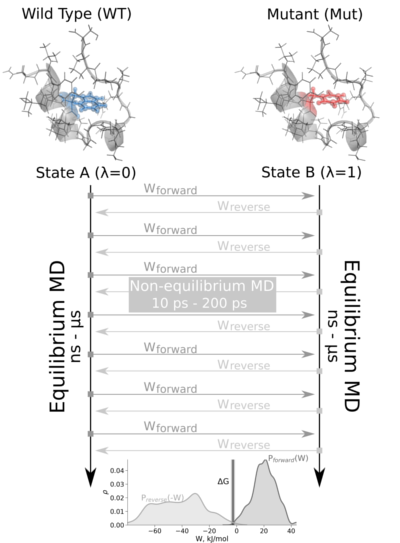
The tutorial calculates the free energy difference in the folded state of a protein. Starting from two 1ns-length independent equilibrium simulations (WT and mutant), snapshots are selected to start fast (50ps) transitions driving the system in the forward (WT to mutant) and reverse (mutant to WT) directions, and the work values required to perform these transitions are collected. With these values, Crooks Gaussian Intersection (CGI), Bennett Acceptance Ratio (BAR) and Jarzynski estimator methods are used to calculate the free energy difference between the two states.
Please note that for the sake of disk space this tutorial is using 1ns-length equilibrium trajectories, whereas in the original example the equilibrium trajectories used were obtained from 10ns-length simulations.
Copyright & Licensing
This software has been developed in the MMB group at the BSC & IRB for the European BioExcel, funded by the European Commission (EU H2020 823830, EU H2020 675728).
- (c) 2015-2026 Barcelona Supercomputing Center
- (c) 2015-2026 Institute for Research in Biomedicine
Licensed under the Apache License 2.0, see the file LICENSE for details.
![]()
Version History
Version 8 (latest) Created 24th Mar 2026 at 15:04 by Genís Bayarri
Update to BioBB 5.2.*
Frozen
 Version-8
Version-86beea68
Version 7 Created 6th Aug 2025 at 09:42 by Genís Bayarri
Update to BioBB 4.2.*
Frozen
Version-7
bc79de5
Version 6 Created 4th Mar 2024 at 14:26 by Genís Bayarri
Update to BioBB 4.1.*
Frozen
Version-6
3706360
Version 5 Created 26th Jul 2023 at 10:23 by Genís Bayarri
Fixed importlib_metadata bug
Frozen
Version-5
098c09d
Version 4 Created 16th Sep 2022 at 10:35 by Genís Bayarri
Update to BioBB 3.8.*. taken from Git commit 5b20069
Frozen
Version-4
a9f9ac3
Version 3 Created 1st Jul 2021 at 11:33 by Douglas Lowe
Updated to BioBB 3.6.0. Taken from Git commit f329e83
Frozen
Version-3
8659eeb
Version 2 Created 7th May 2021 at 14:22 by Douglas Lowe
Updated for GROMACS 2020.4 and BioExcel Building Blocks version 3.5. Taken from Git commit ba3c37d
Frozen
 master
masterf14839e
Version 1 (earliest) Created 15th Sep 2020 at 12:56 by Douglas Lowe
Initial commit. Taken from Git commit 3272c52
Frozen
master
d778945
 Creators and Submitter
Creators and SubmitterViews: 14616 Downloads: 5115
Created: 15th Sep 2020 at 12:56
Last updated: 24th Mar 2026 at 15:04
TagsThis item has not yet been tagged.
AttributionsNone
Collections Interactive Jupyter...
Interactive Jupyter...
Related items
 https://orcid.org/0000-0003-0513-0288
https://orcid.org/0000-0003-0513-0288
BioExcel is the leading European Centre of Excellence for Computational Biomolecular Research. Established in 2015, the centre has grown into a major research and innovation hub for scientific computing. BioExcel develops some of the most popular applications for modelling and simulations of biomolecular systems. A broad range of additional pre-/post-processing tools are integrated with the core applications within user-friendly workflows and container solutions.
The software stack comes with ...
Teams: BioBB Building Blocks, BioExcel Best Practice Guides
Web page: https://bioexcel.eu/

The BioExcel Building Blocks (biobb) software library is a collection of Python wrappers on top of popular biomolecular simulation tools. This library offers a layer of interoperability between the wrapped tools, which make them compatible and prepared to be directly interconnected to build complex biomolecular workflows. The building blocks can be used in many different workflow systems, including Galaxy, CWL, Jupyter Notebook and PyCOMPSs – notably their ...
Space: BioExcel
Public web page: https://mmb.irbbarcelona.org/biobb/

Interactive Jupyter Notebooks in combination with Conda environments can be used to generate FAIR (Findable, Accessible, Interoperable and Reusable/Reproducible) biomolecular simulation workflows. The interactive programming code accompanied by documentation, and the possibility to inspect intermediate results with versatile graphical charts and data visualization is very helpful, especially in iterative processes, where parameters might be adjusted to a particular system of interest. This work ...
Selection of BioExcel Building Blocks (BioBB) Workflows intended for tutorials and training.